7.6 近似原子軌道 − STO
ここでは,原子の一電子波動関数(原子軌道)の近似関数として広く用いられている Slater(スレーター)型原子軌道(Slater-Type Orbital = STO)について述べる。量子化学計算の対象の多くは分子であり,後で述べるように分子の一電子波動関数(分子軌道)は原子軌道の一次結合で近似する。一般に分子の計算では,計算量を少なくするため,原子軌道に含まれる原子ごとのパラメータ(例えばこの節で述べる Slater exponent)を分子を変えるたびに変分法で最適化することはせず,固定値を用いることが多い。
Hartree 方程式
|
(7.6.1) |
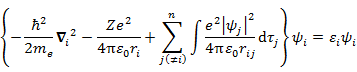
の Hamiltonian のポテンシャル項
|
(7.6.2) |
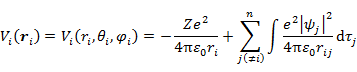
は本来球対称(電子 i の距離 ri のみに依存)ではないが,問題を簡単にするため球対称とおいてみる。そうすると,(7.6.1) は
|
(7.6.3) |
となり,水素類似原子の場合と同じように変数分離が可能になる(5.3 節参照)。
|
(7.6.4) |
(7.6.4) の中の球面調和関数 Y(θi,φi) は水素類似原子の場合と同じであるが,動径分布関数 R(ri) は Vi(ri) がクーロンポテンシャル型ではないため,正確な解が求められない。
Slater は R を水素類似原子の場合と類似した関数で近似することを提案した。すなわち,水素類似原子の R は
|
(7.6.5) |
で表されるが(c は規格化定数),この中の Laguerre の多項式 ![]() を r の最高次数(n − l − 1 次)の項のみに置き換えて,改めて規格化を行うようにする。
を r の最高次数(n − l − 1 次)の項のみに置き換えて,改めて規格化を行うようにする。
|
(7.6.6) |
さらに,(7.6.5) の中に現れる主量子数 n と核電荷 Z を,それぞれ有効主量子数 n*,有効核電荷 Z* に置き換え,Slater exponent を (7.6.7) のように定義する。
|
(7.6.7) |
以上の操作を行うと,Slater の R は次の (7.6.8) の形になる(c' は規格化定数)。
|
(7.6.8) |
(7.6.8) からわかるように,RSlater は方位量子数 l には依存せず,有効主量子数 n* のみに依存している。
Slater は実験値などをもとに,有効主量子数 n*,有効核電荷 Z* を次のように定めた(Slater’s rule)。
|
(1) n* は次表によって定める。
(2) Z* = Z − s とし,s(しゃへい定数とよぶ)は次の手順で定める。
|
1s 軌道(n* = 1)を例にとると,水素原子では ζ = 1,炭素原子では Z* = Z − s = 6 − 0.30 = 5.70 なので ζ = 5.70 と計算される(7.7 節末尾の表参照)。
n* が 1〜3 のとき,RSlater について規格化を行うと,一般式は
|
(7.6.9) |
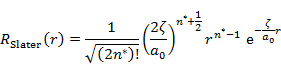
となる。具体的な関数の形は以下のとおりである。
|
(7.6.10) |
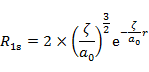
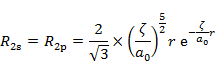
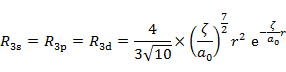
Slaterの R の形は有効主量子数 n* のみに依存し,n* → n,Z* → Z の置き換えを行うと,(7.6.10) の関数の形は,それぞれ水素類似原子の R1s,R2p,R3d の形に等しいことがわかる(5.5 節参照)。
球面調和関数 Y は水素類似原子と共通なので,Slater 型波動関数(Slater 型原子軌道 = STO)の外形は水素類似原子の波動関数と同じである(5.7 節参照)。下の表では,r を原子単位(長さの単位を a0 とする → ζ / a0 を ζ と置くのに等しい)で表し,主量子数 n,方位量子数 l が共通のグループ内で異なる部分を赤字で示している。
| n | l | |m| | ψSTO | |
| 1 | 0 | 0 | 1s | |
| 2 | 0 | 0 | 2s | |
| 2 | 1 | 0 | 2pz | |
| 1 | 2px | |||
| 2py | ||||
| 3 | 0 | 0 | 3s | |
| 3 | 1 | 0 | 3pz | |
| 1 | 3px | |||
| 3py | ||||
| 3 | 2 | 0 | 3dz2 | |
| 1 | 3dxz | |||
| 3dyz | ||||
| 2 | 3dx2-y2 | |||
| 3dxy | ||||
Revised: 2007-08-03
Laguerre の多項式 ![]() の具体的な式は 5.3 節を参照。Laguerre の多項式の最高次数の項は一般に
の具体的な式は 5.3 節を参照。Laguerre の多項式の最高次数の項は一般に
![]()
である。