0.1 分子のエネルギーを求めるときの考え方(概要)
|
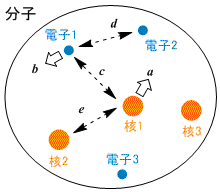
分子のエネルギー |
|
|
核の運動エネルギー = 並進運動 + 回転運動 + 振動運動
|
電子の運動は核の運動に比べて非常に速いので,分子の電子的な性質を考えるときは核が静止していると見なして良い = Born-Oppenheimer 近似
分子の電子エネルギー = |
電子の運動エネルギー Te |
分子の電子エネルギーを求める道筋
-
水素類似原子(1つの核,1つの電子) Te + Vne
Schrödinger 方程式が厳密に解け,原子軌道(AO)とエネルギーが求まる
-
一般の原子(1つの核,2つ以上の電子) Te + Vne + Vee
一電子近似(Hartree-Fock 近似)の導入
一電子波動関数(原子軌道,AO)は水素類似原子の AO その他の関数で近似 一般の分子(2つ以上の核,2つ以上の電子) Te + Vne + Vee + Vnn
一電子近似を適用
原子の AO に代わるものとして分子軌道(MO)を定義
MO は構成原子の AO の線形結合で近似(LCAO-MO 近似)
実際の計算法
そのまままともに計算(いわゆる非経験的分子軌道法における Hartree-Fock 法) |
||
|
近似計算法
|
高精度計算法
|
|
Revised: 2008-05-14